Research Areas in the CMC group
The research within the CMC group focuses on Multiscale-Simulation (MSS) methods for complex Systems, mainly in Materials Chemistry. For the majority of relevant systems, a "brut force" simulation on the level of first principles Quantum Mechanic (QM) methods is impossible from a numeric point of view. In order to tackle the problem, a combination of different methods with different levels of accuracy need to be combined in clever way. This can be done in a sequential fashion (coarser and faster methods are trained on the basis of the finer and more accurate methods) or in concurrent way (different areas are treated with different "resolution" and accuracy at the same time).
As a consequence, our research is always a combination of methodical development and application. We are developing first principles parametrized force fields for metal-organic frameworks (MOFs) and related porous materials. On the "other end" we are working on a real space DFT code for the description of electrified interfaces in contact with an electrolyte (partly funded with RESOLV), using a continuum model to describe the electrolyte.
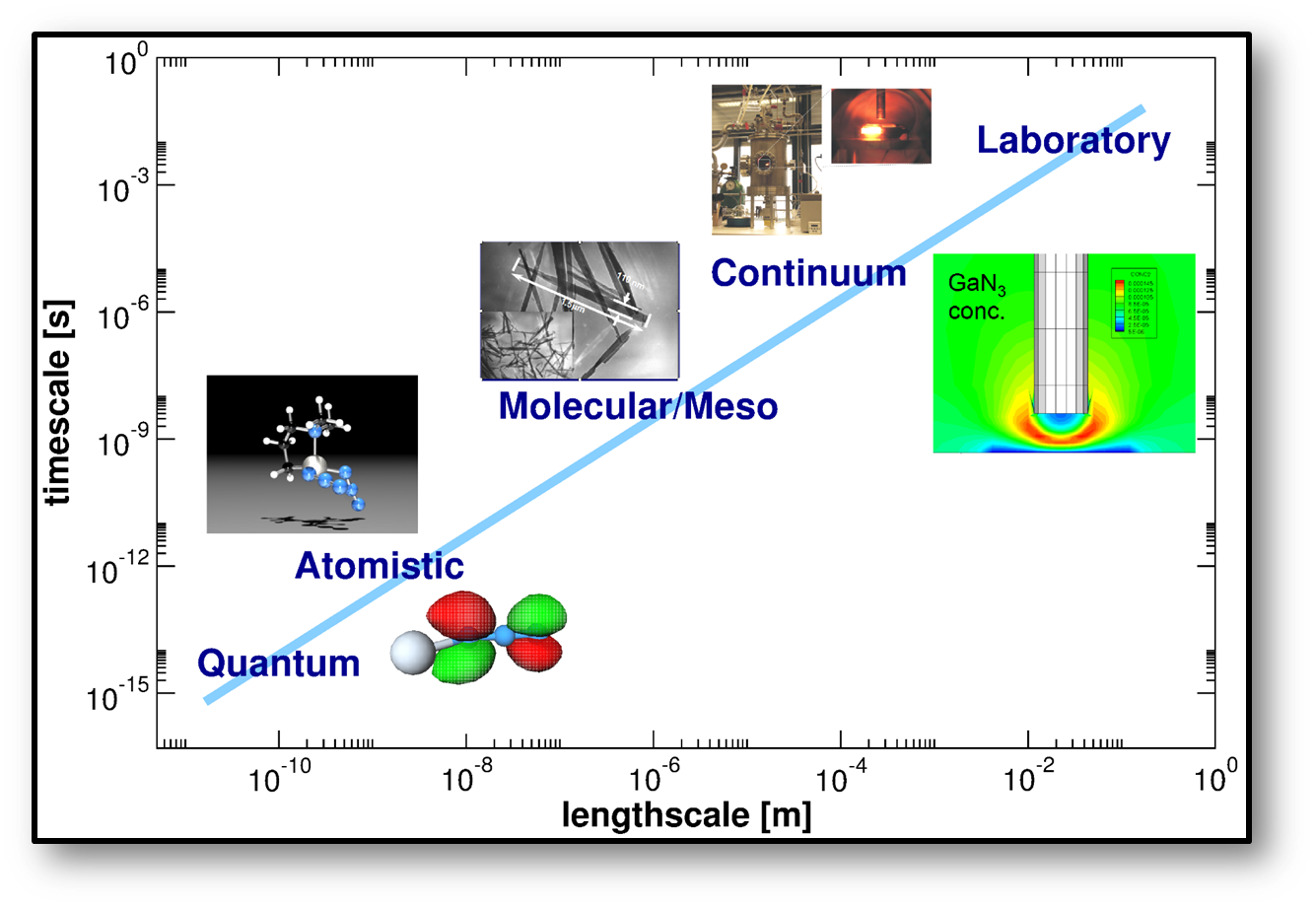
The examples in the above figure are taken from our work on the OMCVD process of the single molecule precursor BAZIGA for the gas phase deposition of GaN. This project was performed in collaboration with Prof. G. Brenner at the TU Claustahl and was funded by the DFG priority program SPP-1119. This project has been terminated.

Methodical Developments
Currently our focus is on development and parametrization of accurate molecular mechanics force fields and on a Car-Parrinello ab initio MD code using real space discretization. (read more)
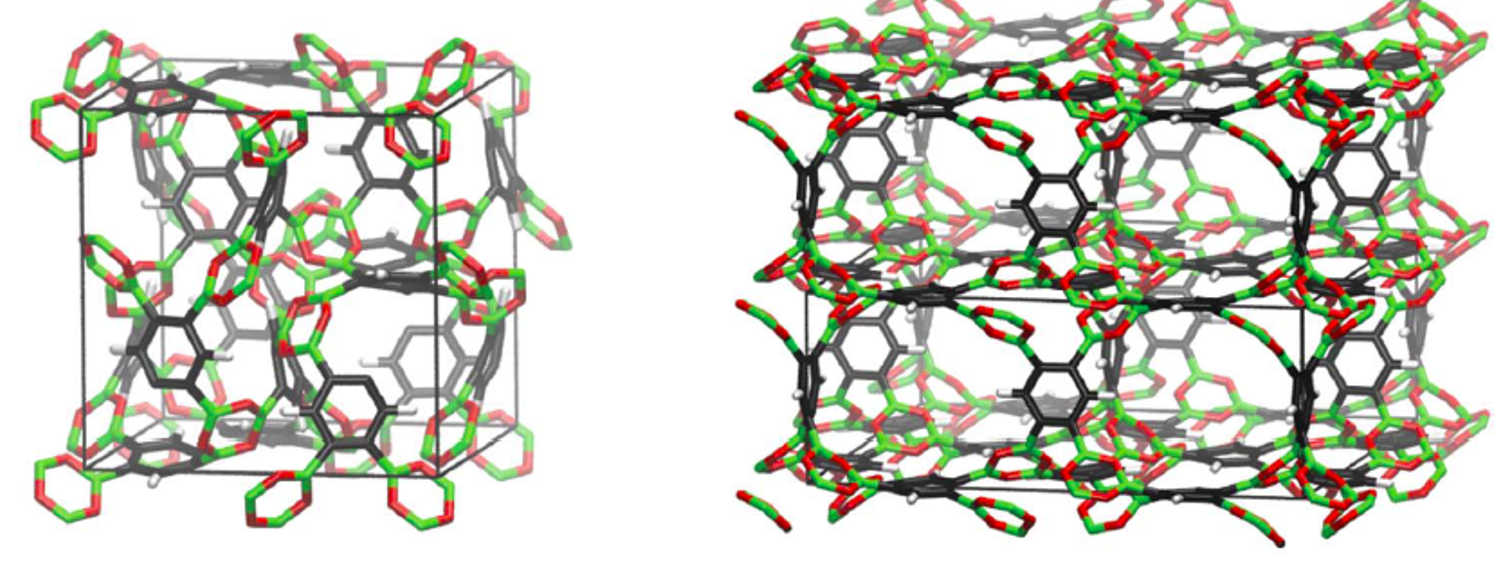
Applications
Our current focus of application is on the simulation of porous MOFs and COFs. Further areas of interest are nano-sized rotors and the simulation of ion solvation and electrolytes at charged surfaces (read more)
