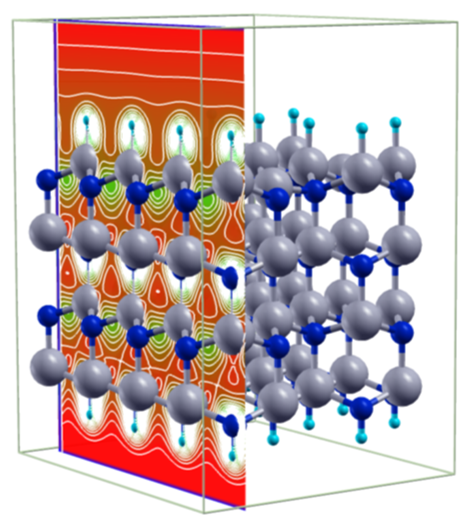
Applications
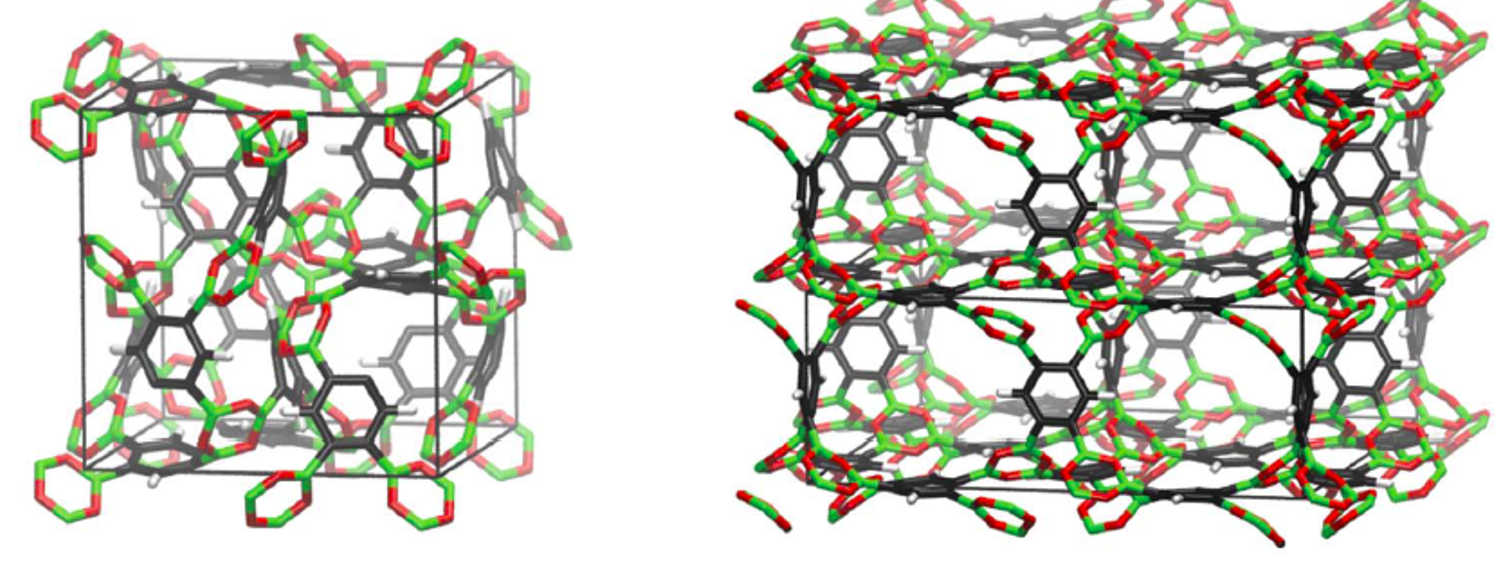
The methods developed in our group can be applied in various areas. However, currently our major focus of application is on the atomistic simulation of porous hybrid materials like MOFs and related materials. We use our first principles derived force field MOF-FF or the simulation of structural and dynamic properties of such systems. The figure below shows two supra-molecular isomers of the related, but purely organic COFs. The hypothetical materials differ only in their network topology but not in their chemical constitution.
Other fields of interest are self-assembled nano-sized molecular rotors, where we also employ our force field MOF-FF. Another area is the simulation of electrochemical systems with our Real Space DFT code. Here the MSS concerns the bridging of length and time scales of sampling the configuration space of the electrolyte.