Dr. Gunnar Schmitz
I develop a computational workflow to automatically determine a heterogeneous reaction mechanism by an automated screening of MD trajectories generated by a reactive force field (ReaxFF) with our findR code. Reaction events are stored in an SQL database and unique reactions are further optimized by electronic structure methods (DFT, Coupled Cluster) to produce rate parameter and thermodynamic data. Currently we are looking to replace the ReaxFF MD step with more accurate semi-empricical methods like the xTB methods. Furthermore a long time goal is to use our methodology as sampling technique for the training of Machine Learning algorithms to construct atomistic potentials.
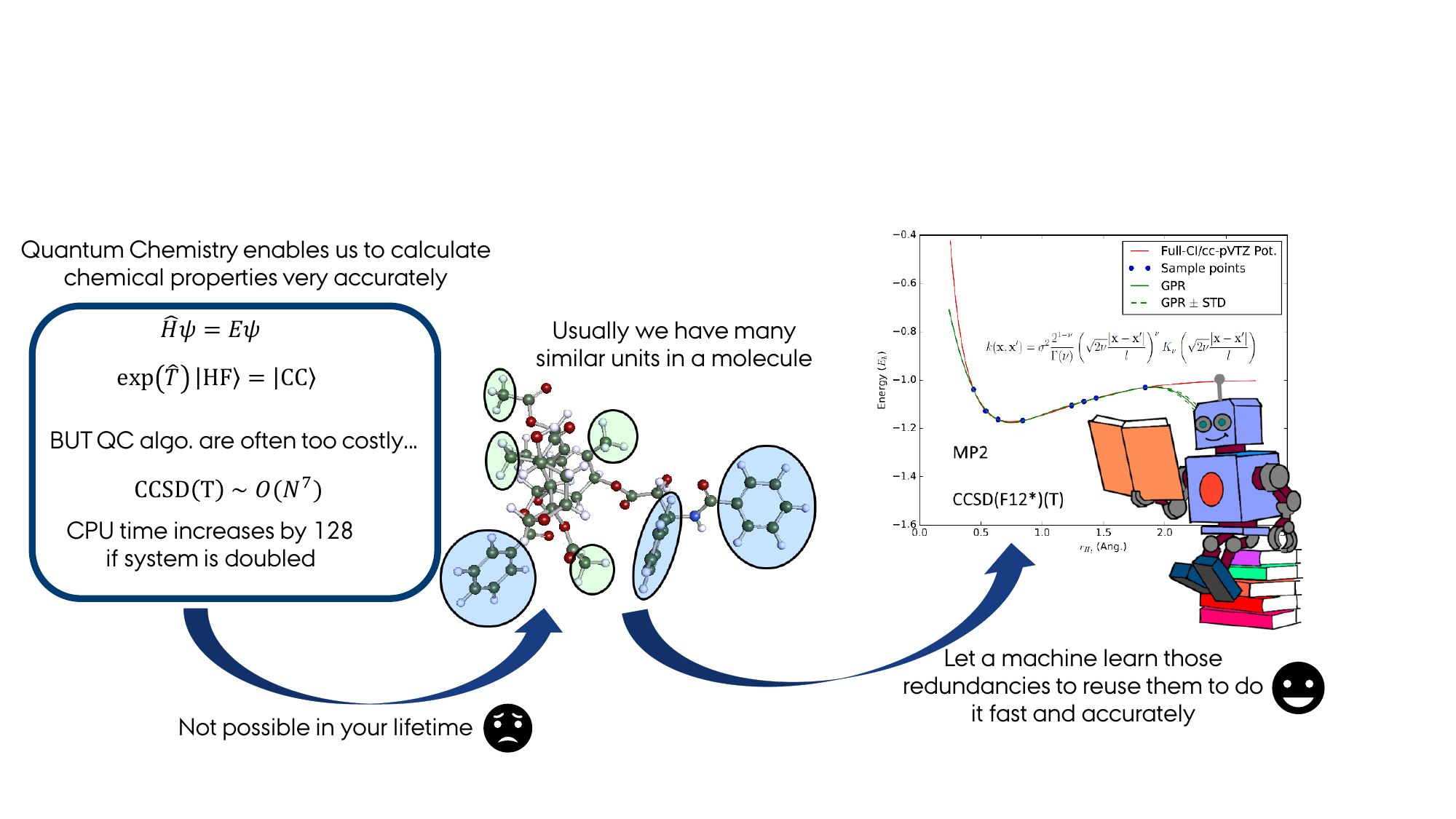
I have a strong background in quantum chemical method development. During my PhD I developed efficient explicitly correlated Coupled Cluster methods using Pair Natural Orbitals. This allowed the calculation of very accurate reaction energies for large molecular systems. After my PhD I changed my focus to the combination of quantum chemical methods with Machine Learning techniques since this allows to bridge even larger scales. My particular expertise lies in using Gaussian Process Regression for on-the-fly generation of potentials.